Introduction
What is NeuroMMSig?
Multimodal Mechanistic Signatures Database for Neurodegenerative Diseases (NeuroMMSig) is designed to allow users to retrieve candidate mechanisms, represented as chains of cause and effect graphs, that fits best to any pattern of experimental data (e.g., gene or SNP set, or a list of imaging features). NeuroMMSig has also been enriched with drug information offering feasible drugs that could be a target for the proposed mechanisms. NeuroMMSig integration of different data scales allows to find the most meaningful mechanisms which suit or better explain the experimental data. This can lead to patient stratification based on data and personilized medicine based on mechanism identification.Introduction
NeuroMMSig offers a web interface where users can submit data to infer mechanistic signatures in the context of neurodegenerative diseases. NeuroMMSig allows submission of multiscale data from molecular to clinical level to return mechanisms that fit best the data. The branding of NeuroMMSig is inspired from Molecular Signatures Database (MSigDB), and the models underlying the server are coded in the Biological Expression Language (BEL).How NeuroMMSig is different from Pathway Databases
- Molecular Profile data is NDD specific: We have curated NDD specific GE data, imaging data etc. On contrast, to general pathway databases are limited to lists of ranked associated pathways (e.g., GSEA) without including disease-related information.
- Multimodal: Multiple types of data is integrated in the knowledge based mechanistic model like genes, chemicals, ions, drugs, imaging and clinical data, SNPs, epigenetics, clinical endpoints etc. On the other hand, pathway databases pathways mostly contain genetic and chemical information.
- Mechanisms are NDD specific: not all available canonical pathways (KEGG, REACTOME etc) are disease specific. Rather it is representing a general static pathway, not a mechanism of a disease. According to KEGG, there are only 10 pathways under NervousSystem Not all genes and relationships described in one canonical pathway may or may not be involving as same as in a disease specific mechanism or some genes are irrelevant to disease context. As an example, figure 1 represents how amyloid beta disrupts the neutrophin signaling pathway in AD via UCHL-1 inhibition and comepting for the binding of NGF with NGFR, which eventually leads to neuron death. On the top of the cartoon, the neutrophin signaling pathway cartoon from Kyoto Encylopedia of Genes and Genomes (KEGG), where some important actors in the example do not appear (Minoru. K, and Goto. S., 2000). More details and examples about this can be found here.
- Tools: Pathway analysis methods do not offer the user the possibility to explore and run algorithms on the networks. However, NeuroMMSig allows to combine and apply different algorithms into the networks. For example, one can merge networks belonging to different disease and compare how a subgraph differs from Alzheimer's to Parkinson's disease. We have also implemented an adapted version of the Network Pertubation Amplitude (NPA). This approach allows users to directly see the effect of expression data on the networks.
What is BEL?
A short introduction to BEL can be find here link. BEL is a language especially designed to represent scientific knowledge in a computable form by capturing causal and correlative relationships in context. In the neurodegenerative disease field, BEL is able to store additional information such as which of relationship exists between the biological entities acting, evidences supporting this relationship in the literature and many other specific annotations such as experiment conditions. Besides, BEL facilitates integration of multiple data types through its flexible and human readable syntax. Therefore, we found BEL ideal to build the models that made the core of NeuroMMSig.
Methodology
The methodology can be grouped into two main sections: Annotation of the mechanistic subgraphs and enrichment ranking algorithm. The first section describes in detail how the manual crafting of the mechanistic subgraphs was performed and the second how the enrichment algorithm works. Both sections are illustrated with examples.
Manual Crafting of the Mechanistic Subgraphs
The aim of the exercise is to generate an inventory of subgraphs, a computable network coded in BEL, that represent and comprise the knowledge about well-established mechanisms or hypotheses involved in the condition. The inventory will support us to better interpret, delineate, and explore the knowledge around disease-specific pathways or mechanisms since we are not looking at the whole vast of knowledge but focusing on a specific part of the disease. Since these subgraphs are computable, they can be merged, modified, enriched, or algorithm can be run on them in order to explore and analyze these hypothese. More detailed information about this part can be found here.
Generating an inventory of disease-related pathways/mechanisms/processes for annotation
First, we needed explore the landscape of disease-related pathways or mechanisms in order to annotate the knowledge assembly with the knowledge from existing terminologies. Having this set of terms also helps us in determining the boundaries of each pathway or mechanism because we can search in the literature whether the the entities in each triplet are involved in each pathway or mechanism. The procedure described next, focuses on the context of Alzheimer's disease (AD) but it can be applied to other conditions (e.g., Parkinson's disease, epilepsy).
- Using SCAIView, we have extracted the Alzheimer's disease related pathways using the query: [MeSH Disease:"Alzheimer Disease"] and show results in the (Pathway Terminology System) 'PTS' terminology. The resulting query extracted pathway terms described in the context of Alzheimer's disease from over 100000 articles in MEDLINE. Following, a list of approximately 900 terms was exported.
-
The next step involved manual curation as well as an enrichment on this primary list. Manual curation was
required address the following issues:
- Remove false positives (e.g., "Melanogenesis")
- Wrong entries (e.g., "No mapping")
- Duplicates (e.g., "amyloidogenesis" and "amyloid-beta peptide pathway")
- Synonyms pathways/processes/mechanisms were grouped together into one consensus term (e.g., "amyloidogenesis" and "amyloid-beta peptide pathway" was labelled as "Amylodogenic pathway").
After collecting the set of terms that are used to describe the pathways and mechanisms in AD, we used this set as an inventory for the annotation of the knowledge assembly together with other pathway repositories and disease specific literature. However, during the curation process, new disease-specific pathways/mechanisms were found while reading literature and inspecting the AD Knowledge Assembly so they were included and curated into the inventory. Therefore, this was an iterative process of curating the inventory parallel to the annotation of the knowledge assembly.
Manual Curation and Annotation of the Knowledge Assembly into Mechanistic Subgraphs
Mapping biological entities to mechanisms
In order to map an entity with its corresponding disease-specific mechanisms (Note that an entity might be part of multiple mechanisms), we would first read the corresponding evidence in order to search for insights about possible mechanisms that might be described in the evidence itself. Next, with the help of the evidence/context we would query the literature to find supporting evidences in the case we have already in mind a mechanism that might be related with the entity or simply search what is its role in the condition. This would involve, for example, using text mining resources or search engines (e.g., SCAIView, PubMed, Google) and querying the entity name (e.g., gene/protein name) together with the name of the condition. The results of the queries will pinpoint publications that describe associations between the entity and the condition (if exists). By reading these documents insights about the role of the entity in pathophysiological mechanisms can be gained.
In this case, PubMed identifiers of the documents can be used as references that support this mapping. If the queries do not provide any insight about possible mechanisms in which the entity might be involved in, dedicated databases (e.g., Reactome, UniProt for proteins, CHEBI for chemicals) can be queried to complement the search. They might provide valuable information about the role of the entity in pathways related with the disease as well as supporting references.
Finally, if this extensive search concludes without identifying mechanistic links with the entity, we would assume that the entity cannot be linked with any mechanism.
It is important to remember that the spectrum of biological scales that are part of NeuroMMSig varies from the chemical space to clinical endpoints. Therefore, it might be arduous or even impossible to link some entity types to mechanisms (e.g., clinical endpoints like brain region volumes). Moreover, the procedure to map entities to mechanisms varies depending on the entity type. For example, in the case of genes we would first investigate databases like UniProt or pathway databases such as Reactome or KEGG. However, investigating the link between a specific lipid and a mechanism might require to look at dedicated databases like CHEBI.
This mapping process is conducted in parallel with the annotation of the BEL document. That means that by annotating every statement, we would try to see whether the entities are linked with any mechanism, and we would proceed with the annotation if links have been found. Furthermore, we would add to a mapping file the references support the link between the entity and the mechanism. Below, some examples of the annotation process.
Annotation examples
Below some examples of annotations of BEL statements
Example 1: This example shows how a simple triplet with its corresponding NeuroMMsig subgraphs was annotated. In this example, we have a triplet containing two entities associated. The first one is a gene (EPHA1) that codes for a protein related to Akt signaling, and the second one is the node representing a condition (Alzheimer's disease). Therefore, we would first check whether the subject or the object are associated with any AD mechanism. For that, we would use text mining resources or search engines (e.g., SCAView, PubMed, Google). After an extensive search, we would conclude that the gene EPHA1 is involved in Akt signaling, a pathway related to AD. Next, we would add the corresponding references that support this link to the mapping file. Finally, we would annotate this BEL statement only with the Akt subgraph since the Alzheimer's disease node is a general entity not associated with any mechanism in particular.
SET Citation = {"PubMed", "XXXX", "XXXXX"}
SET Subgraph = "Akt subgraph"
g(HGNC:EPHA1) association path(MESHD:"Alzheimer Disease")Example 2: This example shows how a triplet that presents a relationship between a chemical and a biological process was annotated. In this case, we would first check in the literature if the chemical corticosteroid or inflammation processes plays a role in the disease. After the search, we concluded that corticosteroid is not involved with any mechanisms known in AD, and inflammation is a well-known process in AD. Therefore, this BEL statement (triplet) is annotated with the "Inflammatory response subgraph", the network that comprises all the knowledge around inflammation processes in the context of AD.
SET Citation = {"PubMed", "XXXX", "XXXXX"}
SET Evidence = "high-dose steroid treatment decreases vascular inflammation and ischemic
tissue damage after myocardial infarction and stroke through direct vascular effects involving the
nontranscriptional activation of eNOS"
SET Species = "9606" #Taxonomy ID- Homo sapiens
SET Tissue = "Vascular System"
SET Disease = "Stroke"
SET Subgraph = "Inflammatory response subgraph"
a(CHEBI:corticosteroid) decreases bp(MESHD:"Inflammation")Example 3: In some cases, it is required to not only investigate the link of the entities in the triplet with a disease-specific mechanism but also the relationships that is part of the triplet. For instance, when amyloid beta protein is not correctly processed and leads to amyloid plaque formation we talk about the "amyloidogenic pathway/process". However, when the amyloid beta protein is processed correctly we talk about the "non-amyloidogenic pathway". Therefore, we annotated the AD Knowledge Assembly using two annotations representing each different pathway depending on the relationship involved in the triplet.
SET Citation = {"PubMed", "XXXX", "XXXXX"}
SET Evidence = "Protein X increases Amyloid Beta 42 fragment"
SET Subgraph = "Amyloidogenic subgraph"
p(HGNC:X) increases p(HGNC:APP, frag(672_713))SET Citation = {"PubMed", "XXXX", "XXXXX"}
SET Evidence = "Protein X inhibits APP"
SET Subgraph = "Non-amyloidogenic subgraph"
p(HGNC:X) decreases p(HGNC:APP)Example 4: The last example shows how triplets might be involved in multiple pathways. Therefore, a triplet (subject-relationship-object) can be annotated to multiple subgraphs as it is shown in the following example where the triplet is part of two different subgraphs (one linking EPHA1 to "Akt subgraph" and the other linking inflammation to its corresponding subgraph).
SET Citation = {"PubMed", "XXXX", "XXXXX"}
SET Evidence = "AKT1 is positively correlated with inflammation processes
SET Subgraph = {"Akt subgraph", "Inflammatory response subgraph"}
p(HGNC:EPHA1) positiveCorrelation bp(MESHD:"Inflammation")Enrichment Ranking Algorithm
The enrichment ranking algorithm allows user to prioritize subgraphs given the enrichment score. Therefore, the user submits their data, the algorithm calculates a score for the data-mapped subgraphs as a way to prioritize further exploration. Following, more details about the algorithm.
The enrichment algorithm evaluates a score given three different scores and their corresponding weights (equation 1). The weights allow the users to bias the algorithm towards some of the measurements more than others. They can be modified from zero to one, but by default they are set to one so each of the three scores have the same weight in the enrichment score.
$$s=w_{1}s_{1}+w_{2}s_{2}+w_{3}s_{3}$$Following, we provide a detailed description of each of the scores.
Proportional number of nodes mapped to the subgraph
In similar manner to pathway analysis methods — Over-representation analysis (ORA) in particular — measuring the percentage of mapped nodes is a common approach to calculate the enrichment of a network (Khatri et al., 2012). The higher the number of data that is mapped to nodes is, the greater the score. It is important to mention that not all nodes in the subgraphs can be mapped. For instance, entity types such as biological processes, chemicals or pathologies cannot be easily associated with mechanisms or pathways. Therefore, since they are not associated with subgraphs, only entities like gene sets and their corresponding associated imaging features and SNPs are taken into consideration when calculating the proportion of nodes that can be mapped.
$$s_{1} = \frac{\#Matches}{\#PossibleMatches}$$Score 1 example: An user submits two genes as an input (DDR2,CRYAB) that map to this imaginary subgraph (graph below). In this case, the score 1 would be 1 since there are only two mappeable nodes (DDR2,CRYAB). The other two nodes (inflammatory response and innate immune response) are biological processes and cannot be mapped with data. If only one of the two genes is submitted, the score would be 0.5 (1/2).
$$s_{1} = \frac{2 (mapped)}{2 (possible mappings)} = 1$$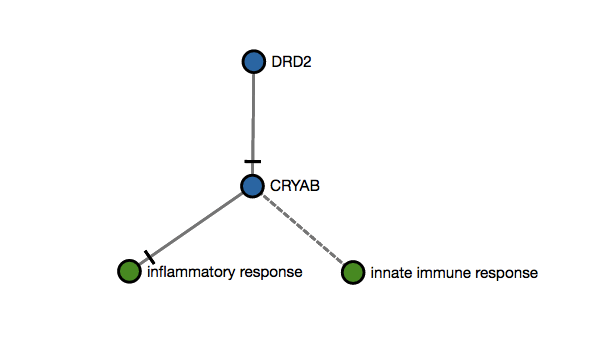
Analysis of graph topology: hubs
The second score aims to assign weights to nodes depending on their contribution to graph topology — following a similar strategy of PT-based methods. We compare different methods described in the literature and betweenness centrality was found a good indicator of node importance since it reflects the amount of control that this node exerts over the interactions of other nodes in the network. This approach of measuring node importance in biological networks coincided with other studies in the literature (Gu et al., 2012 and Joy et al., 2005).
NeuroMMSig considers then the top 5% of nodes in each subgraph with highest betweenness centrality as hubs. The calculations were performed removing all pathology nodes. Since pathology nodes are highly connected in the networks, their presence distorted shortest paths calculations which are the basic principle of betweenness centrality. When a network was smaller than 20 nodes, there is no ‘node-weighing’ based on hubs. As a conclusion, when data is mapped to any of these hub nodes, enrichment score for its correspoding subgraph is increased by the proportion of mapped hubs. As an example, if a network has two hubs and one of them is mapped, the value of the score accounting for hubs is 0.5.
Score 2 example
An user submits two genes as an input (DDR2,CRYAB) that map to this imaginary subgraph (graph below). Let's suppose that the betweenness centrality analysis in this subgraph ranks the CRYAB as the only hub. Then, score 2 would be 1 since there is only one hub and it has been mapped (1/n when is the total number of hubs in that subgraph). Please note that this is a simple illustration that would not apply since NeuroMMSig only calculates this score for graphs over 20 nodes (top 5% nodes are considered as hubs).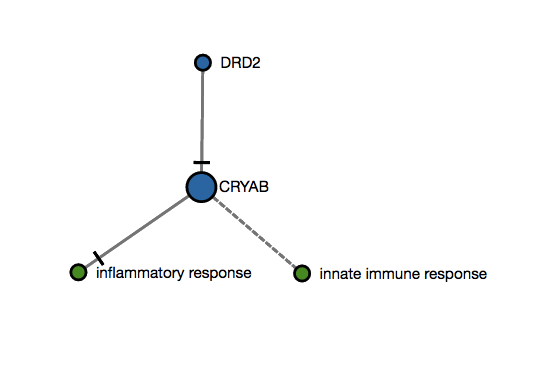
Analysis of graph topology: connectivity
The last score accounting for the final enrichment score is based on whether the nodes that have been mapped are close to each other and thus, their connectivity. Since mechanisms are chain of cause and effects (a collection of nodes and edges in subgraphs) one would expect a better enrichment score for a series of mapped nodes that are closely together than series where nodes are disperse between them. Therefore, this score assigns higher scores to neighbour nodes rather than spread ones. For that we used equation 2, where NG[i] is the number of mapped nodes that are also neighbours of node i and n is the total number of nodes mapped. Here, it is important to remark that because BEL implies directionality, networks are considered as directed graphs. As a consequence, when calculating this score from node 1 to node 2 (1->2), node 1 is a neighbour of 2 but not the other way around because one cannot traverse from node 2 to 1 in a direct graph. The denominator n(n-1) intends to normalize the score to 1 since n(n-1) are the total of possible connections in a directed graph.
It is important to mention that we do not consider BEL graphs multi-edge property. Thus, there would be no difference in the score if they are multiple edges going from node 1 to node 2, or only one edge. Summarizing, the score only considers the presence or not of an edge between nodes, not the type/number of them.
$$s_{3} = \frac{\sum_{i}^{n} N_{G}[i]}{n(n-1)}$$Equation 2. Analysis of graph topology: connectivityScore 3 example
An user submits two genes as an input (DDR2,CRYAB) that map to this imaginary subgraph (graph below). In this subgraph, both genes are connected with one edge (DDR2 decreases CRYAB). In this case, the score 3 would be 0.5, since there is only one edge between the mapped nodes and the total of possible edges between them is 2 (n=2). $$s_{3} = \frac{1}{2(2-1)} = 0.5$$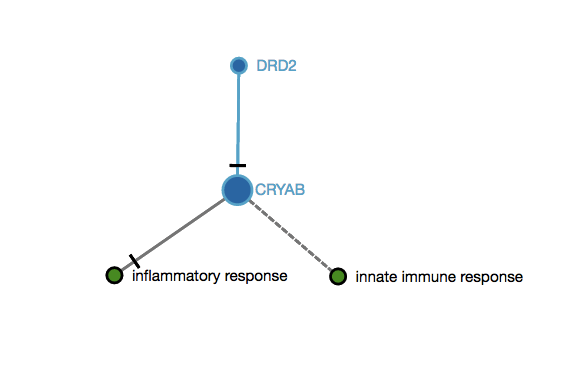 The final score when submitting these two genes (DDR2,CRYAB) taking into consideration the examples
above would be:
$$s=w_{1}1+w_{2}1+w_{3}0.5$$
The final score when submitting these two genes (DDR2,CRYAB) taking into consideration the examples
above would be:
$$s=w_{1}1+w_{2}1+w_{3}0.5$$
Mechanisms in subgraphs:
What do we call a mechanism?
“A chain of causes and effects forms a pathophysiological context, where minor dysregulation of molecular events may aggregate at a network level and lead to a pathological deviation from the normal state (Hofmann-Apitius et al., 2015)".
Once data is mapped to the subgraphs, we can identify the different ways the data-mapped nodes dysrupt a particular node of interest such a biological process. For more detail about how NeuroMMSig might identify possible dysregulated paths in the networks, please visit "How to use NeuroMMSig" section.
References:
Gu, Z. et al. (2012) Centrality-based pathway enrichment: a systematic approach for finding significant pathways dominated by key genes. BMC systems biology 6.1: 56.
Joy, M. P. et al. (2005) High-betweenness proteins in the yeast protein interaction network. BioMed Research International. 2: 96-103
Kanehisa, Minoru, and Susumu Goto. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28.1 : 27-30.
Khatri, P. et al. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol 8.2 e1002375
Kodamullil, A. et al. (2015) Computable cause-and-effect models of healthy and Alzheimer's disease states and their mechanistic differential analysis. Alzheimer's & Dementia 11.11 : 1329-1339.
Martin Hofmann-Apitius et al. (2015) Bioinformatics Mining and Modeling Methods for the Identification of Disease Mechanisms in Neurodegenerative Disorders. eng. In: Int J Mol Sci 16.12, pp. 29179–29206. doi: 10.3390/ijms161226148. url: http://dx.doi.org/10.3390/ijms161226148